

 EN
ENCEP是Certification of suitability to the monographs of the European Pharmacopoeia的缩写。CEP是用于阐述某原料的质量符合欧洲药典相关章节的要求,以及符合在CEP中载明的其他补充测试。
尽管CEP不是在欧洲国家范围内用于药品生产的原料获得监管批准的强制性要求,然而,根据活性成分质量部分的申报文件指南的声明1,CEP是首选选项。
同时,CEP也是用于阐明TSE风险的法规符合性首选形式2。

在2024年5月,EDQM发布了CEP的《Content of the dossier for sterile substances》草案,公开征求意见,针对无菌原料的申请者为获取CEP,给出了一系列的文件要求。无菌生产工艺中的一系列信息需要在3.2.S.2.5章节中提交。
灭菌方法的依据
大部分情况下,无菌原料是通过除菌过滤后(采用最大标称孔径0.22μm)灌装到已灭菌的容器中。也有采用干热灭菌、电离辐射、环氧乙烷灭菌形式,这几种方法需要根据指南的要求进行详细的论证和说明。
生产区域
从污染控制策略(Contamination Control Strategy, CCS)角度,无菌原料的生产应该在合适的洁净室中。目前,针对微生物和颗粒物的风险控制,需要选择合适的生产设备,如RABS、隔离器或其他系统。从本质上,生产区域的选择需要EU GMP附录1的要求。3
除菌过滤/无菌工艺的生产工艺汇总
汇总的信息内容包含溶剂、温度、设备、预过滤和除菌过滤、结晶、晶种、离心、分离、体积缩小、亚批次的混合、冻干、干燥、灌装等,还需在CEP中明确批次大小。
使用的过滤器信息
CEP要求提供使用的过滤器的详细信息,包含过滤器材质、标称孔径、过滤器数量、过滤面积,还需包含过滤条件和参数(如,最大过滤时间、最大过滤批量、最长过滤器使用周期、最长生产周期、操作压力,等)。
过滤器验证
针对除菌过滤器,验证的内容需要有证明能达到除菌级别的微生物挑战测试数据、吸附、化学兼容性、可提取物/浸出物数据以及对应的独行风险评价。
过滤器和工艺设备的灭菌
相关的灭菌需要符合EU GMP附录1的要求。4
预过滤生物负载
跟全球范围内的除菌过滤前的溶液微生物负载要求保持一致,即最终除菌过滤前的溶液微生物负载水平不能超过10 CFU/100 mL。除菌过滤的时间一般不超过一个工作日(24小时),如果采用更长的过滤时间,需要有数据充分支持。在原液的制备后和最终除菌过滤前的最大间隔时间,也需要在工艺中明确,并且有数据支持该最大间隔时间不会影响工艺中微生物的负载水平。
过滤器的重复使用
当原料的生产存在过滤器的重复使用,需要做好相应的重复使用的评估和验证工作,用以降低交叉污染、内毒素超标、兼容性、除菌性能等风险。这些风险的引入,可能是来自于清洗、存放条件、重复灭菌、外部污染、材料耐受性等方面。
无菌工艺
无菌工艺的时间尽量缩短,用以降低潜在微生物污染的风险。原液的保持时间以及最终灌装的时间需要明确,如果超过24小时,就需要进行充分的风险评估。
工艺模拟/验证
需要识别生产中的无菌工艺步骤,进行连续三批培养基灌装试验确认工艺操作的无菌保障。接受标准应当是没有微生物生长。根据最新的EU GMP附录1的要求,每年应当进行两次工艺模拟测试(大约每六个月一次)。
包装灭菌
原料的包装材料灭菌应当按照相关的要求进行,目前有两个标准可参考Ph. Eur., 5.1.1和EMA/CHMP/CVMP/QWP/8503745。同时,包装材料的完整性需要进行验证。
复检间隔
申请者需要复检时,在间隔时间终点时,稳定性的测试研究需要采用商用化包装材料一致的包装,也应当涵盖无菌测试。
总而言之,EDQM在对无菌原料的CEP申请时,其关注的核心内容之一即无菌工艺的保障。当采用除菌过滤的工艺,过滤工艺的设计显得尤为重要。尤其是在目前EU GMP附录1针对除菌工艺需要按照能进行使用前灭菌后完整性测试(即,Pre-use post sterilisation integrity test,PUPSIT)。
乐纯生物在针对过滤工艺设计和过滤器的产品应用有着丰富的经验:
LeSiever®PES筒式 / 囊式过滤器,采用除菌级亲水性聚醚砜(PES)膜,兼具高流速和高载量的两大优点。
LeSiever®PES筒式 / 囊式过滤器卓越的化学稳定性和耐高温性能,经过验证的可靠细菌截留能力,可广泛应用在细胞培养、终端制剂、原液等相关工艺的降生物负荷和除菌过滤,是生物制药领域的理想之选。

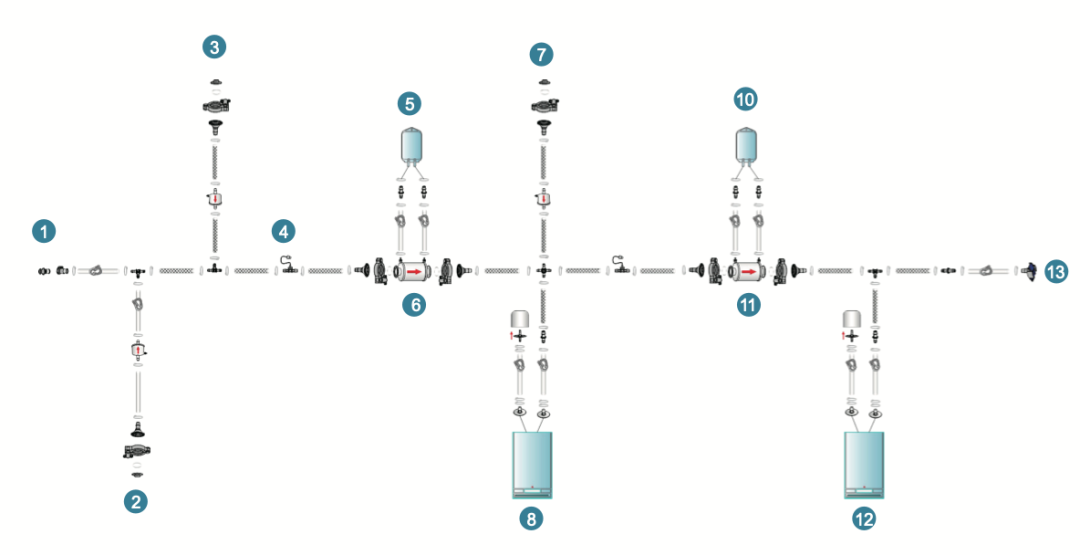
乐纯生物根据不同地区的法规要求,设计了不同的冗余过滤系统,PUPSIT版冗余过滤采用水润湿不吹干。
- 亲水滤器,用于除菌滤器的润湿
- 空气滤器,用于冗余过滤系统完整性检测
- 润洗水的接收容器
- 蝶式气体滤器,用于排除废液袋中气体
产品优势:
- 灵活定制
- 供应稳定
- 超净生产平台,有效控制颗粒异物
- 100%完整性测试,ready to use
长按识别/扫描上方二维码
企微直连,获取服务

乐纯生物集团
产品专员
参考文献:
1.Note for Guidance on summary requirements for active substances in the quality part of the dossier (CPMP /QWP/297/97 Rev 1 corr; EMEA/CVMP/1069/02)】,
2.general chapter 5.2.8 of Ph. Eur. = EMA NfG 410/01 on minimising the risk of transmitting animal spongiform encephalopathy agents via medicinal products】。
3.Annex 1 of of Eudralex Volume 4 EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use】
4.Annex 1 of of Eudralex Volume 4 EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use
5.Ph. Eur., 5.1.1 Methods of preparation of sterile products;EMA/CHMP/CVMP/QWP/850374,Guideline on the sterilisation of the medicinal product, active substance, excipient and primary container】。

