

 EN
EN在生物制药领域,无菌过滤工艺是确保最终产品质量和安全性的关键环节之一。其中,用于去除细菌和控制生物负荷的除菌过滤系统一直是无菌过滤工艺中的重要环节。终端除菌过滤组装设计和PUPSIT操作实施中,具体有哪些核心点,需要充分考量呢?
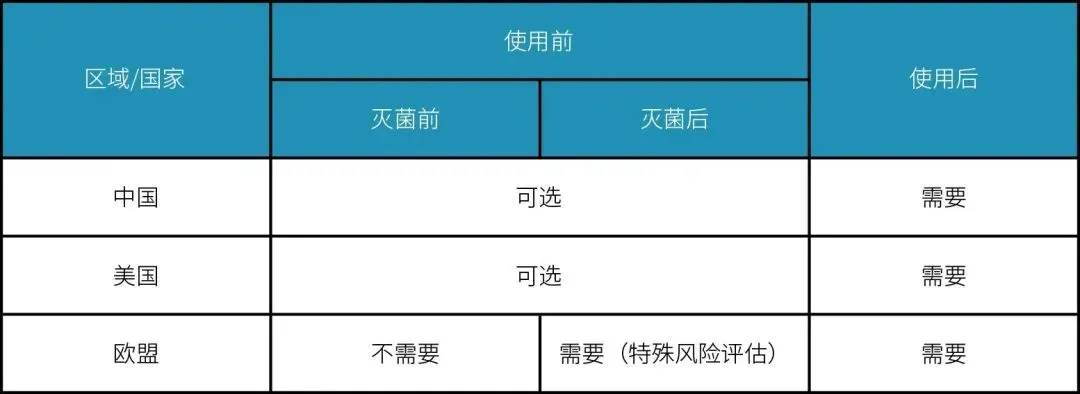
在生物药的生产过程中,药品除菌最常用的方法是使用除菌级过滤器。不同国家和地区对除菌级过滤器的完整性有不同的要求,其中以欧盟 GMP的要求最为严格,即在药品生产的终端过滤步骤中,要求PUPSIT(pre-use post sterilisation integrity test)。
我国GMP对PUPSIT的要求是基于对药品生产过程的关键环节进行验证,保证生产过程的稳定性和可控性。
推荐阅读:除菌过滤器PUPSIT实施要点及风险考量
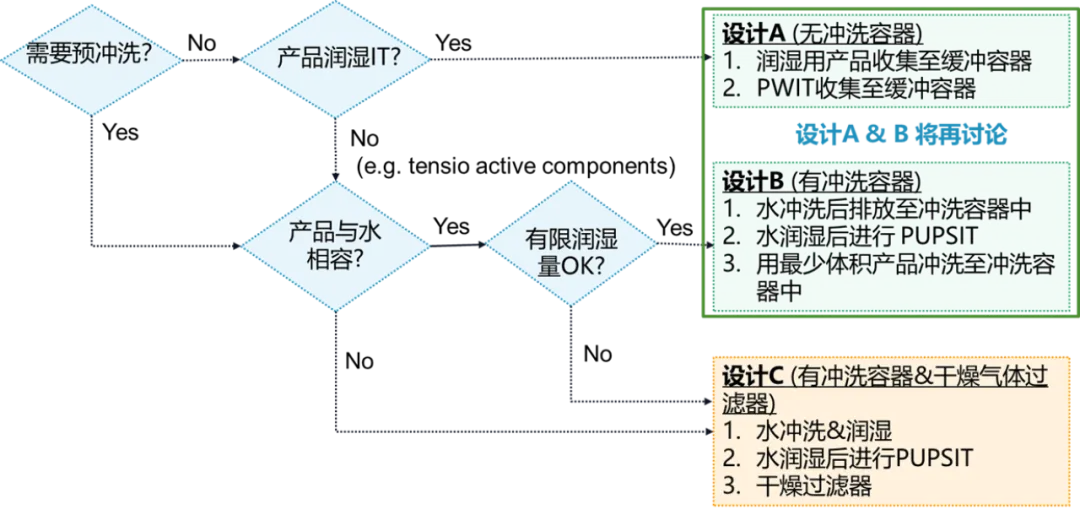
冗余过滤装置本身可降低产品污染风险。乐纯生物根据不同地区的法规要求,可设计不同的冗余过滤系统,在设计前需要充分考虑:
- 完整性检测润湿液体:料液直接润湿完整性、水润湿完整性
- 滤器是否需要吹干:气体滤器大小、气体滤器是否耐受伽马辐照
- 泄露测试:管路种类(耐压管路/非耐压管路)、袋子与管路是否一起检测
- 废液袋大小:滤器面积大小/润湿体积及速度/重复冲洗/重复PUPSIT/安全余量,滤器Hold-Up Volume
PUPSIT过滤系统设计决策树
完整性检测法规总结
- EMA, Good Manufacturing Practice, Annex 1: Manufacture of Sterile Medicinal Products , 2008
- WHO Technical Report Series, No 961, Annex 6, 2011
- India FDA, Good Manufacturing Practices and Requirements of Premises, Plant and Equipment for Pharmaceutical Products, 2005
- Japan, Manufacture of Sterile Pharmaceutical Products by Aseptic Processing, 2006
- Korea, Manufacture of Sterile Medicinal Products, 2010
- TGA, Australian Code of Good Manufacturing Practice for Medical Products, 2002
点击查看大图
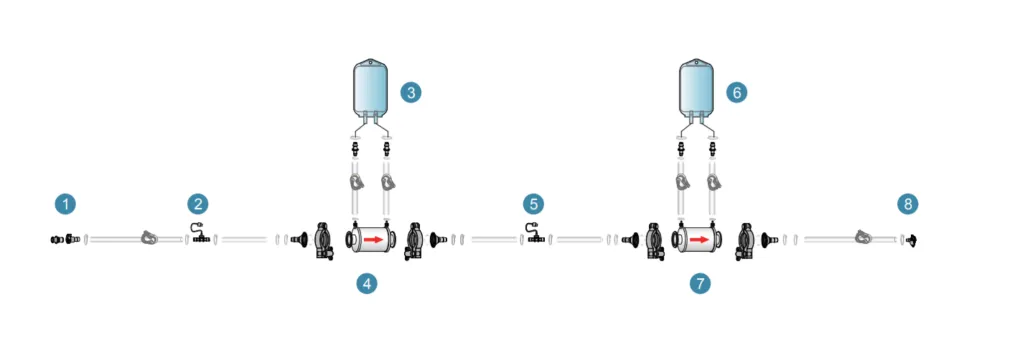
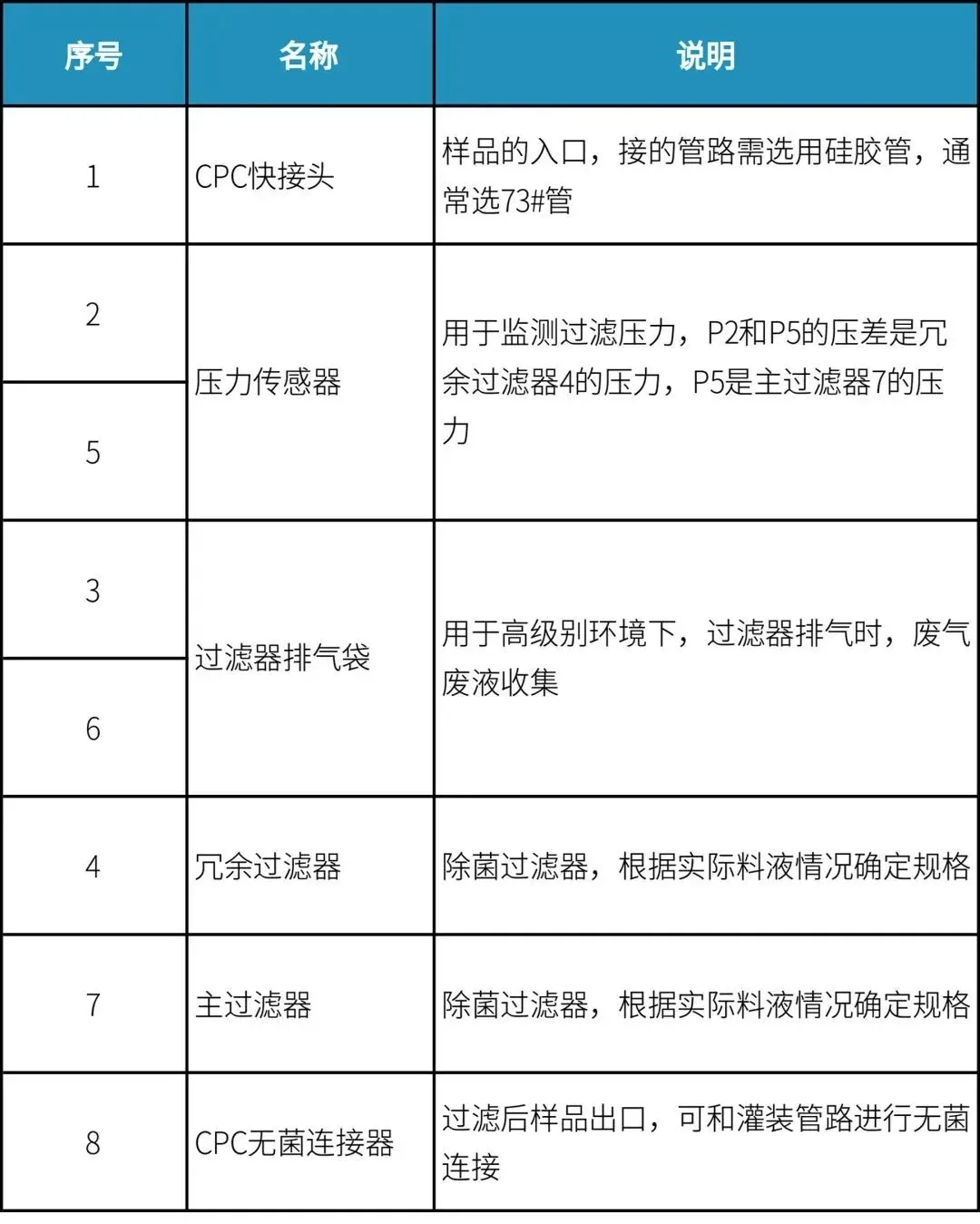
终端过滤组装1(非PUPSIT)
设计考量要点如下表,仅供参考:
点击查看大图
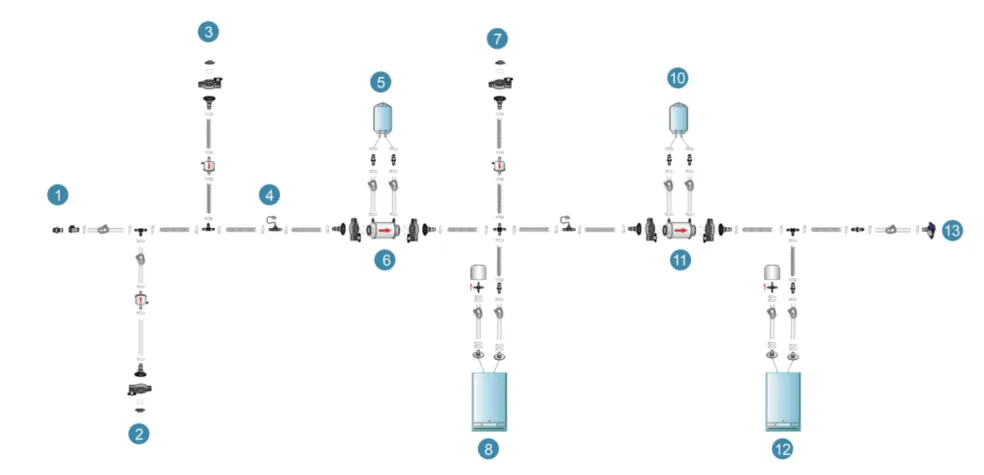
终端过滤组装2(PUPSIT,水润湿不吹干)
设计考量要点如下表,仅供参考:
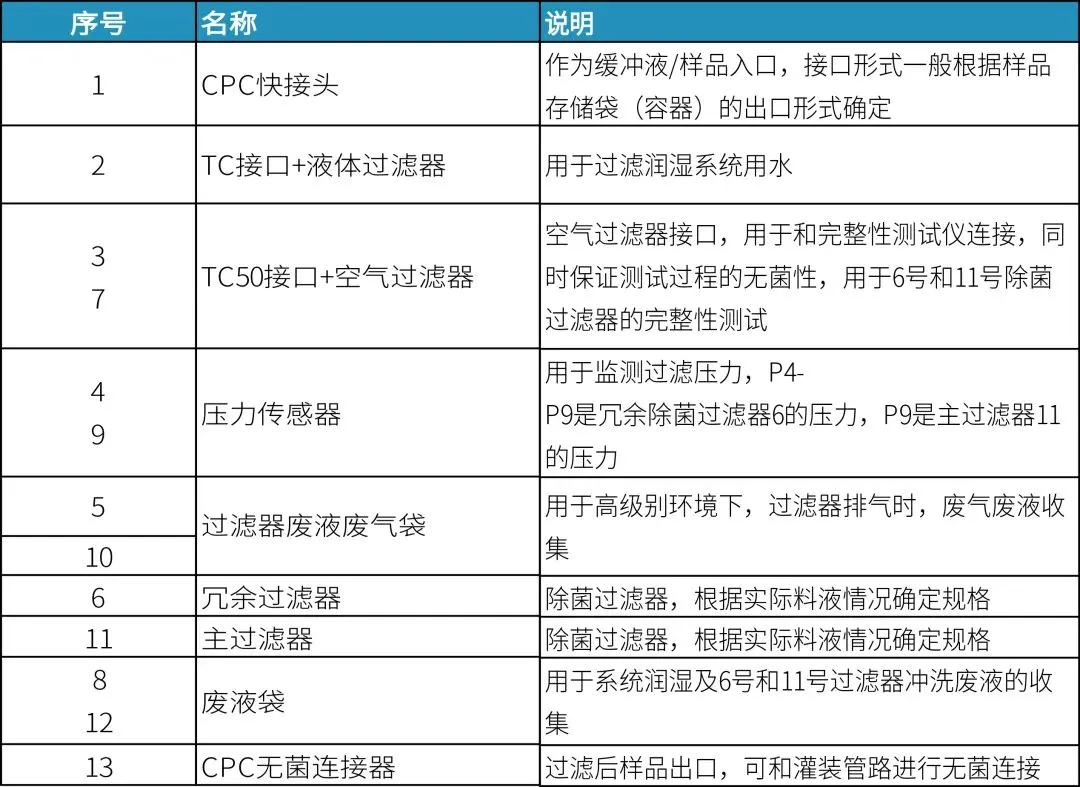
点击查看大图
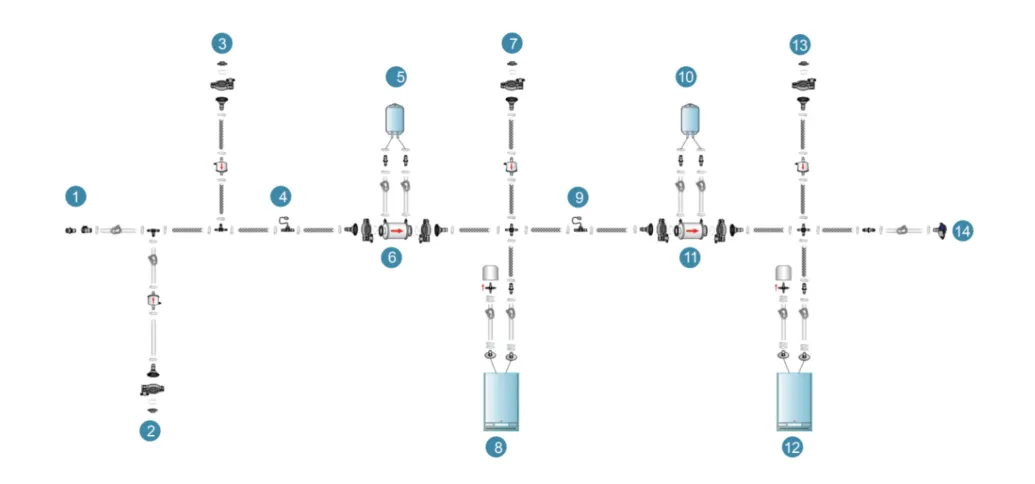
终端过滤组装3(PUPSIT,水润湿,吹干)
设计考量要点如下表,仅供参考:
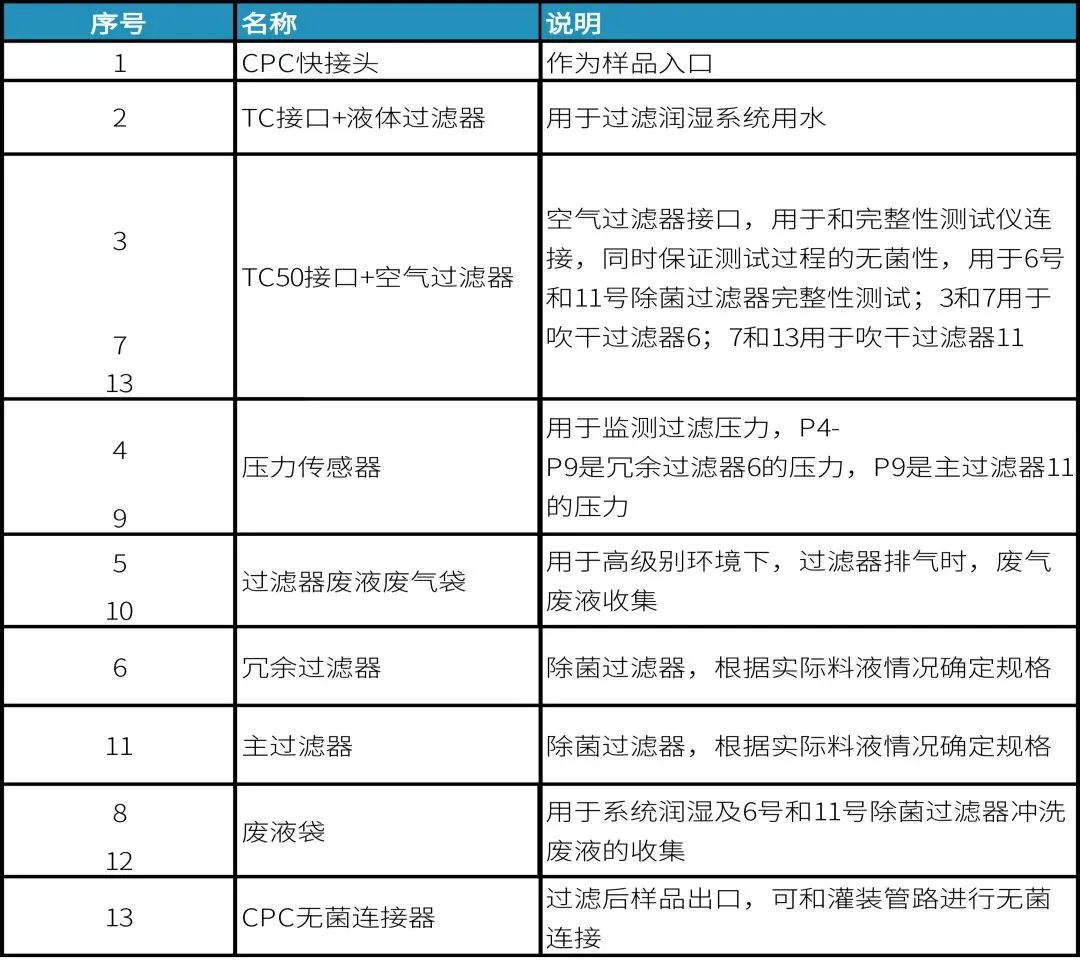
点击查看大图
在此附上关于PUPSIT实际操作步骤(未完整版)
(视频)
如想获取完整视频,请长按识别/扫描如下二维码
与我们产品专员进行咨询联系

乐纯生物集团
产品专员
作为生物药GMP生产的最后环节,无菌制剂灌装需要专业的知识、良好的设计及严格的规定,以保证最终成品的质量。生物药的完整制剂生产工艺包括:原液准备及冻融、原液混匀、除菌过滤、无菌灌装、加塞、(冻干)、轧盖、灯检、放行检测、贴签/包装等过程。

乐纯生物深耕生物制药行业,作为国内一次性耗材的先行企业,有着丰富的一次性管路组装设计经验。为制药企业提供快速、高效的解决方案。
生物制药蓬勃发展的今天,一次性技术为制药生产带来巨大变革的潜力,它能降低新药成本、改善生产灵活性、减少循环时间、避免清洁验证、缩短建立生产工艺所需时间,节省生产运营总成本。
